在药物研发、蛋白构象研究、核酸功能解析等领域,分子模拟是理解生命过程的重要工具,但长期面临“尺度-精度”的根本矛盾:高精度的量子化学方法通常只能处理数百原子规模的体系,而更具扩展性的经典力场又难以准确刻画复杂的分子相互作用与非局部效应。尽管机器学习力场(MLFF)的兴起显著提升了模拟精度,但在真实的生物大分子场景中,依然常常被三类问题制约:公开数据集多以小分子为主,缺少包含显式溶剂的大型体系样本;长程相互作用难以被完整建模;以及大规模体系下的推理吞吐不足,难以真正融入稳定可用的工程化流程。
近日,IQuest Research(至知创新研究院)UBio团队正式推出面向生物大分子体系的高精度模拟框架 UBio‑MolFM。该框架通过数据、模型架构与训练策略的协同设计,试图在分子基础模型领域实现新的突破。团队为此构建了迄今为止最大的高精度DFT数据集 UBio-Mol26,包含超过1700万条数据,体系最大可达1200个原子,并重点覆盖了溶液环境中的蛋白质、DNA/RNA片段、细胞膜等真实生物大分子。在此海量数据基础上,团队采用了先进的 E2FormerV2(一种类Transformer的等变神经网络架构),结合对长程相互作用的显式建模,并在多阶段课程学习框架的加持下,实现了SOTA级别的精度与吞吐表现。与此同时,团队开源了500万条高质量DFT数据子集 UBio‑Protein26 5M,以推动社区复现与应用,后续模型权重和推理代码也将陆续公开。
本次发布:UBio-MolFM框架、Protein26数据底座与开源复现路径
UBio‑MolFM 是一套面向真实生物体系场景的分子基础模型框架,旨在覆盖蛋白质、DNA/RNA、脂质膜及其复合物体系,并针对显式溶剂、长程耦合与大体系稳定推理进行了系统性设计。整个框架包含三个核心部分:一是面向生物体系的数据底座(UBio-Mol26/Protein26);二是用于大体系高效推理的等变架构实现(E2Former-V2);三是支持多理论层级稳定融合的训练范式,并配套了开放的代码、数据与后续工作计划。
数据底座:更接近真实生物体系
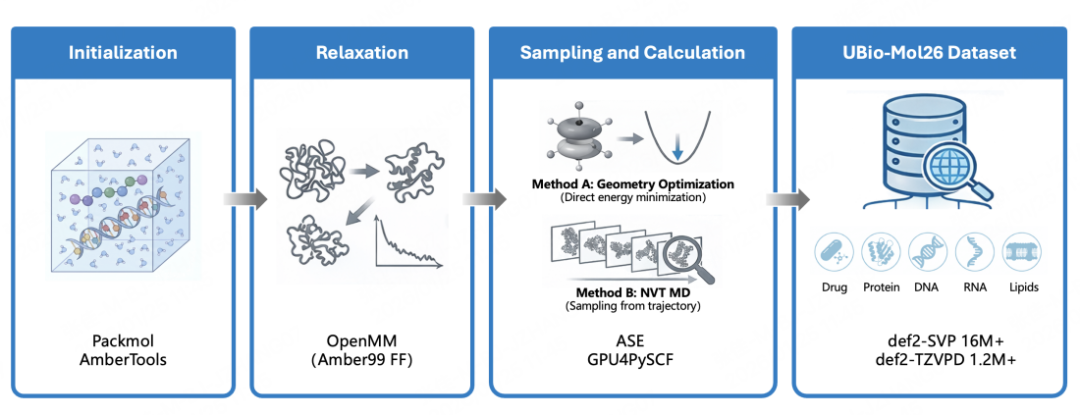
数据是模型性能的基石。UBio-Mol26 数据集规模约1700万构型,覆盖了蛋白质、DNA/RNA、脂质膜与复合体系,包含显式溶剂环境,单体系原子数最高可达1200个,并采用统一流程生成高质量的DFT标注(使用wB97M-D3泛函)。同时,团队开放了标准化的子集 UBio-Protein26 5M(500万训练 + 20万测试),其中训练集由450万条def2-SVP基组数据和50万条def2-TZVPD基组数据组成。该子集的平均原子数超过370,有效弥补了公开数据在生物大体系尺度上的覆盖空白。
在数据构造策略上,UBio-Mol26 采用了“自下而上枚举”与“自上而下采样”相结合的混合路线:
- 自下而上:系统枚举氨基酸短肽、核酸片段与脂质单元,确保基础分子构建块的广泛覆盖。
- 自上而下:从真实的蛋白质结构中抽取局部环境,并进行溶剂化与化学封端处理,以增强对生物场景中复杂几何与相互作用模式的刻画能力。
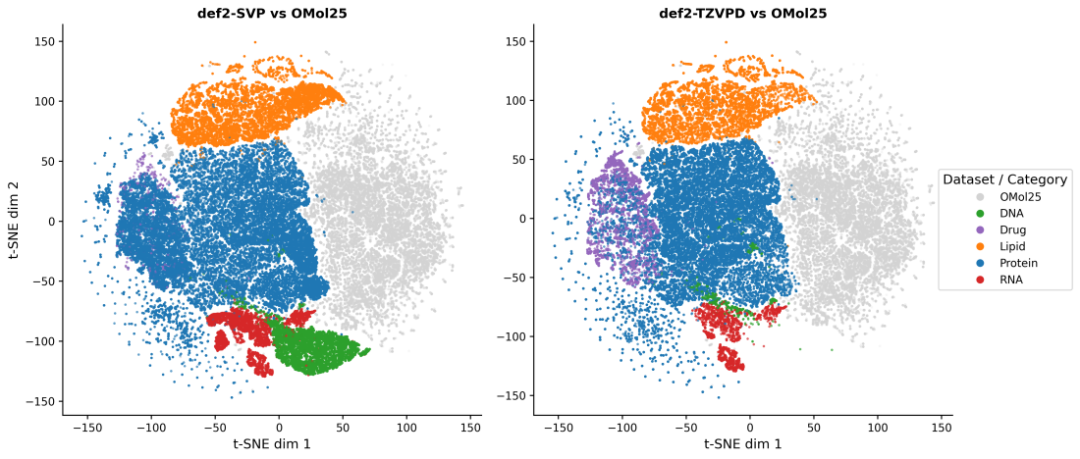
团队还通过t-SNE降维可视化,对比了UBio-Mol26与现有知名小分子数据集OMol25在特征空间中的分布,直观展示了两套数据间的互补关系。
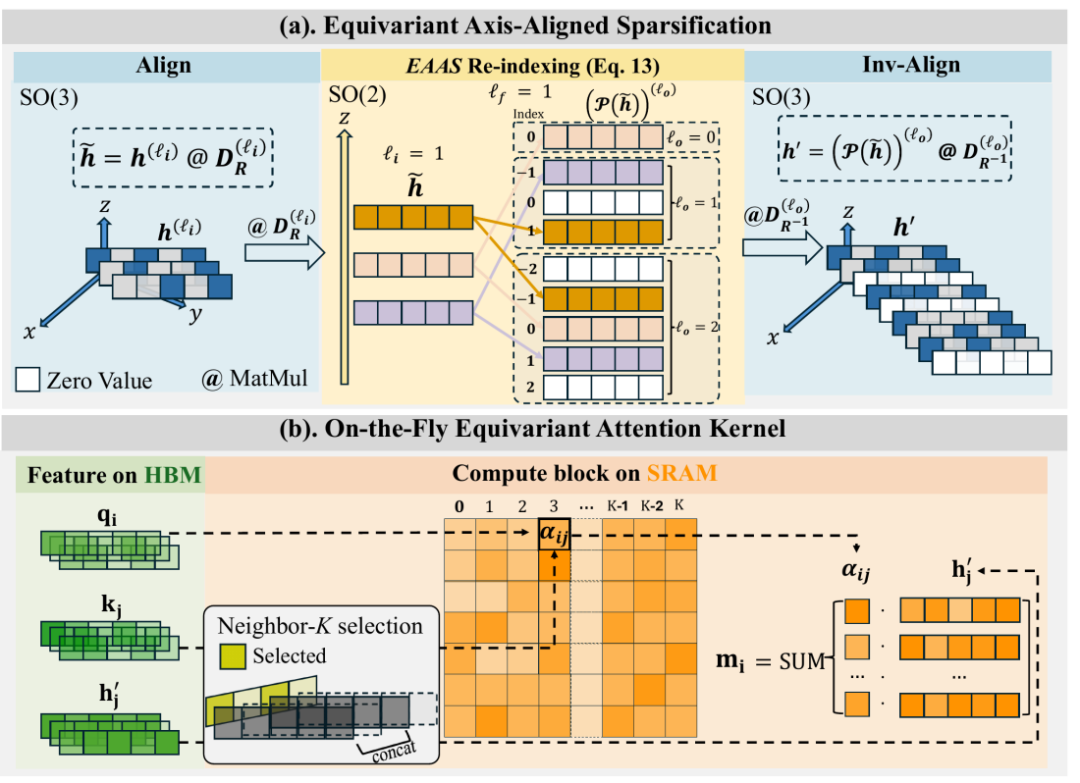
UBio-MolFM 采用了 E2Former-V2 架构,这是一种设计上更注重硬件友好性的等变Transformer。其以“节点为中心”的实现方式减少了稀疏图上的操作开销,提升了内存访问的局部性,从而有效降低了大体系推理时的计算成本。同时,模型通过等变轴对齐稀疏化(EAAS) 技术降低了SO(3)群张量积的计算复杂度,并结合长短程(LSR)建模模块,能够同时高效处理局部相互作用与远程耦合效应。这些对于Transformer架构在大规模科学计算中的应用是关键的优化。
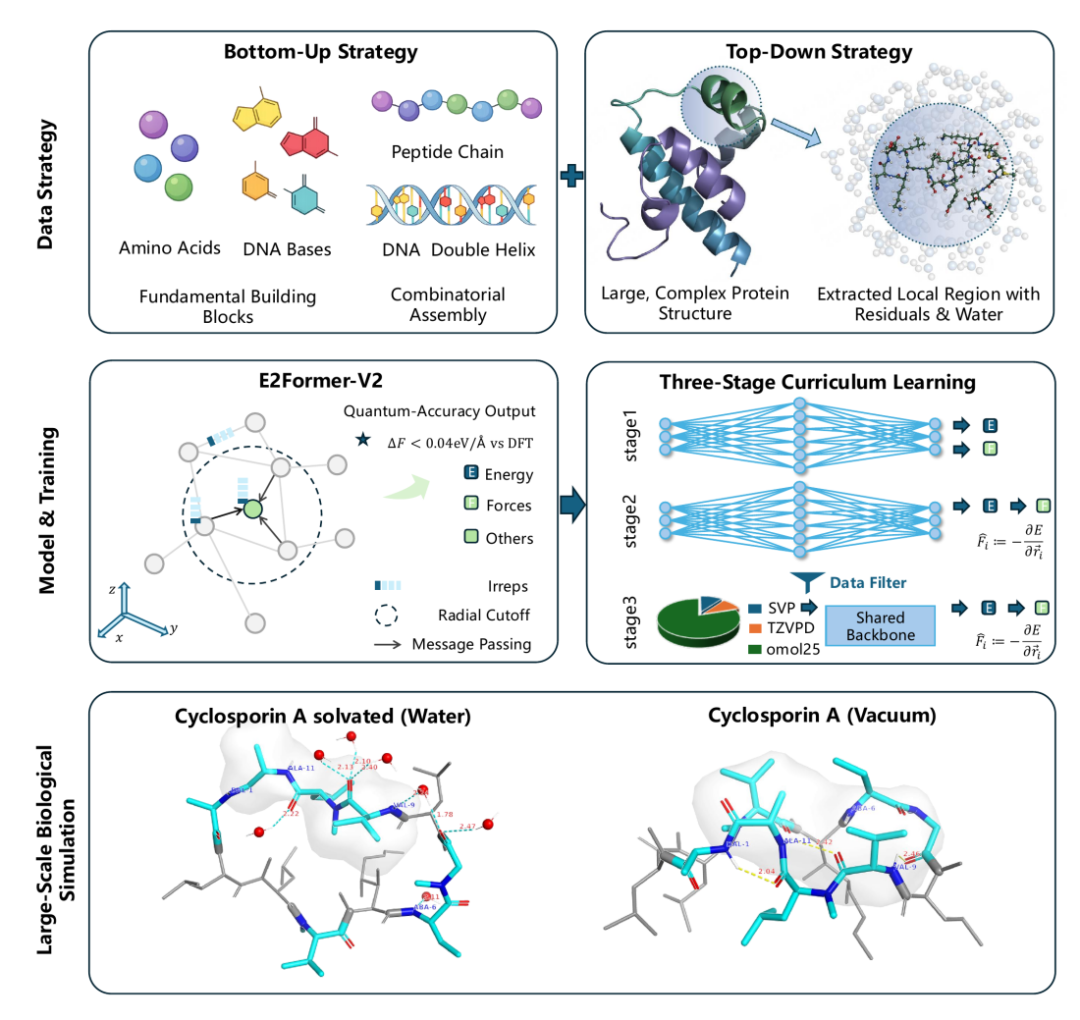
训练:三阶段课程学习(稳定融合多理论层级)
为了在化学多样性覆盖、物理一致性约束与体系尺度扩展之间取得平衡,团队设计了三阶段课程学习策略:
- 阶段一:使用OMol25数据集进行快速的能量初始化,采用能量头与独立的力头并行预测,以提升训练吞吐量。
- 阶段二:丢弃独立的力头,力完全由能量梯度自动计算得到,以此强化能量与力之间的物理一致性。
- 阶段三:融合UBio‑Mol26的多精度数据。通过双头结构处理不同理论层级的数据:SVP和TZVPD数据分别对应不同的能量预测头,其中TZVPD数据仅施加力损失监督,以绕过不同基组间可能存在的系统性能量偏移。同时,采用OMol25、SVP、TZVPD以8:1:1的比例进行数据混合,并辅以相似性过滤,以保持训练过程的稳定性。
在生物大体系上同时实现“可引用精度”与“大规模吞吐”
对于科研与产业应用而言,一个分子基础模型要真正融入日常工作流,必须回答两个核心问题:第一,在更接近真实的生物大体系上,其预测误差是否仍然可控且可验证?第二,在需要长时间轨迹与高频推理的分子动力学场景中,其吞吐量是否足以支撑工程化使用?UBio-MolFM 的价值在于,它试图在同一套评测与工程约束下,并行推进这两方面的能力。
外推精度:在1,300–1,500原子规模上实现显著领先
团队构建了一个专门用于外推测试的数据集,体系规模在1,300至1,500个原子之间,并在此测试集上对比了 MACE-OMol 与 UMA-S-1p1(均使用官方代码与检查点,评测设置与官方保持一致)。该测试集覆盖了蛋白质优化、DNA优化、RNA优化、蛋白质MD等多种任务。
| 数据类型 |
样本数 |
轨迹数 |
平均原子数 |
| 蛋白质优化 |
1,010 |
10 |
1,524.9 |
| DNA 优化 |
226 |
5 |
1,289.6 |
| RNA 优化 |
505 |
5 |
1,467.4 |
| 蛋白质 MD |
875 |
18 |
1,434.6 |
在代表性的蛋白质优化任务中,UBio-MolFM (S3) 的相对能量平均绝对误差(MAE)为 8.68 meV/100 atoms,显著优于 MACE-OMol 的 76.94 和 UMA-S-1p1 的 83.45;其力的 MAE 为 16.77 meV/Å,也明显低于对比模型的39.29和42.84。
| 模型 |
相对能量 MAE |
力 MAE |
ΔE MAE |
| MACE-OMol |
76.94 |
39.29 |
1.62 |
| UMA-S-1p1 |
83.45 |
42.84 |
2.22 |
| UBio-MolFM (S3) |
8.68 |
16.77 |
0.83 |
团队评估指出,在蛋白质相关任务上,模型在能量与力的误差方面均有显著降低;而在DNA任务上仍有提升空间,这已被明确为后续数据扩展的重点方向。
物理一致性:验证从溶剂结构到金属配位的关键细节
在生物体系中,许多看似细微的物理量,恰恰决定了模型能否用于可靠的机理解释与预测。UBio-MolFM 在多个层面进行了物理一致性验证:
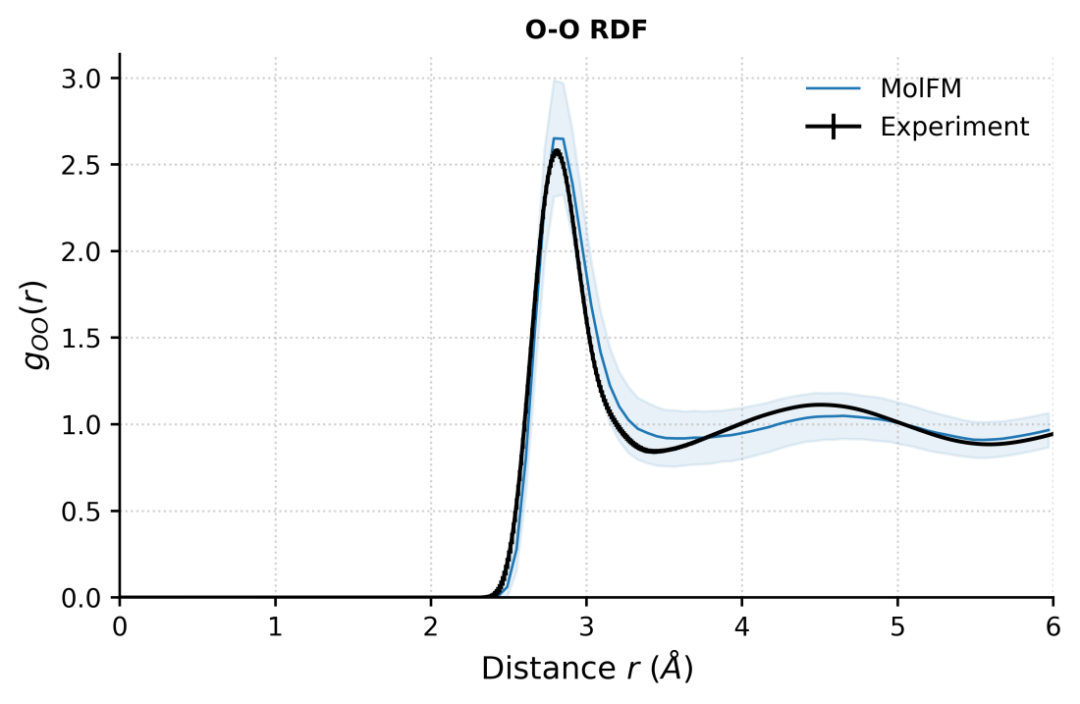
- 溶剂结构:在纯水与0.15 mol/L NaCl溶液中,模型能够重现径向分布函数(RDF)的结构特征与配位数,验证了其对基本液体结构的合理刻画。
- 环境敏感构象:在环孢素A(CsA)体系中,模型能够正确保持其环境敏感的构象状态——在水溶液中维持开放态,在真空中维持闭合态,这表明模型对溶剂化环境变化的物理响应符合直觉。
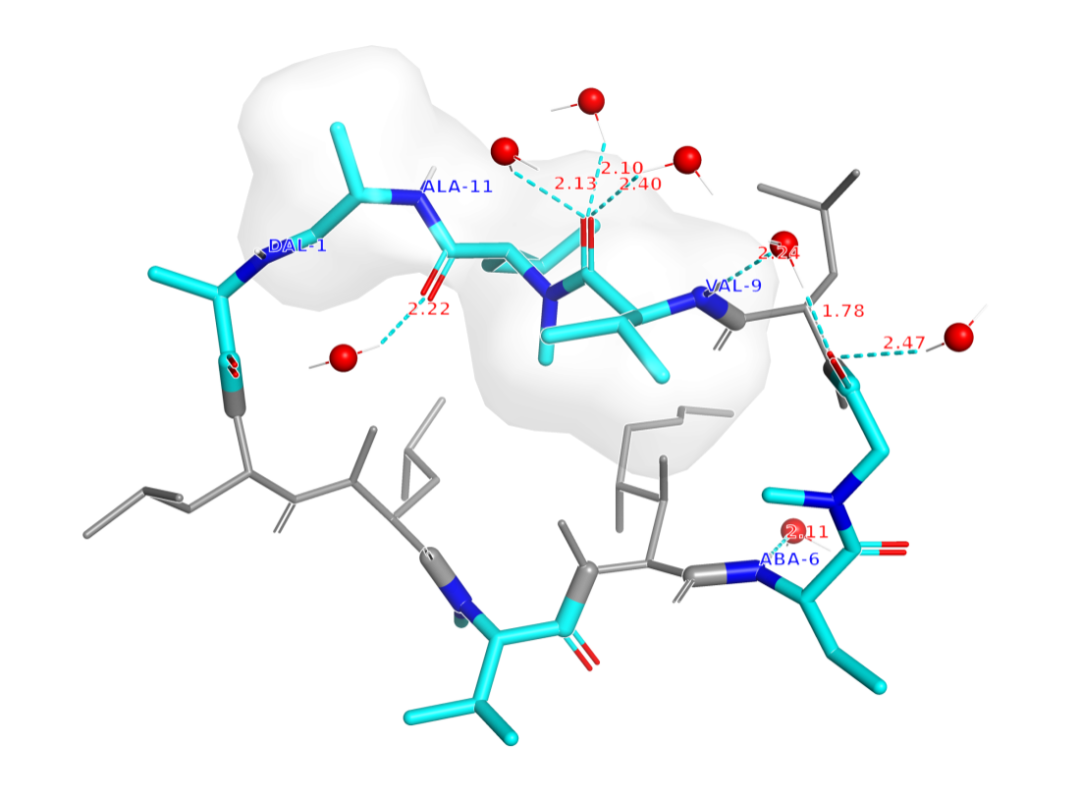
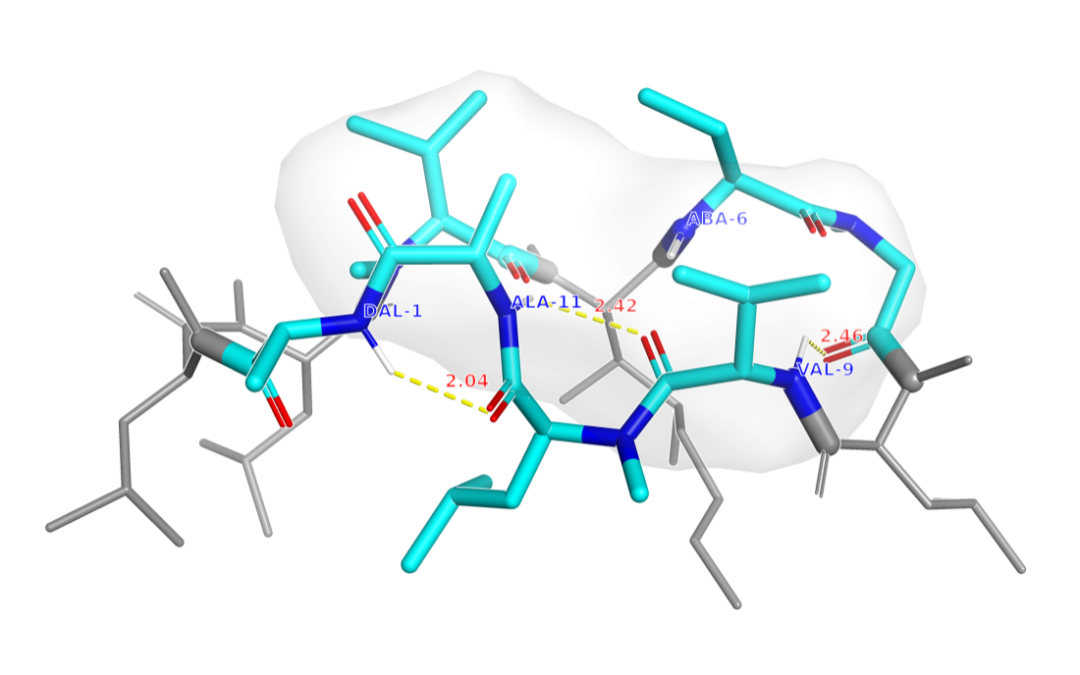
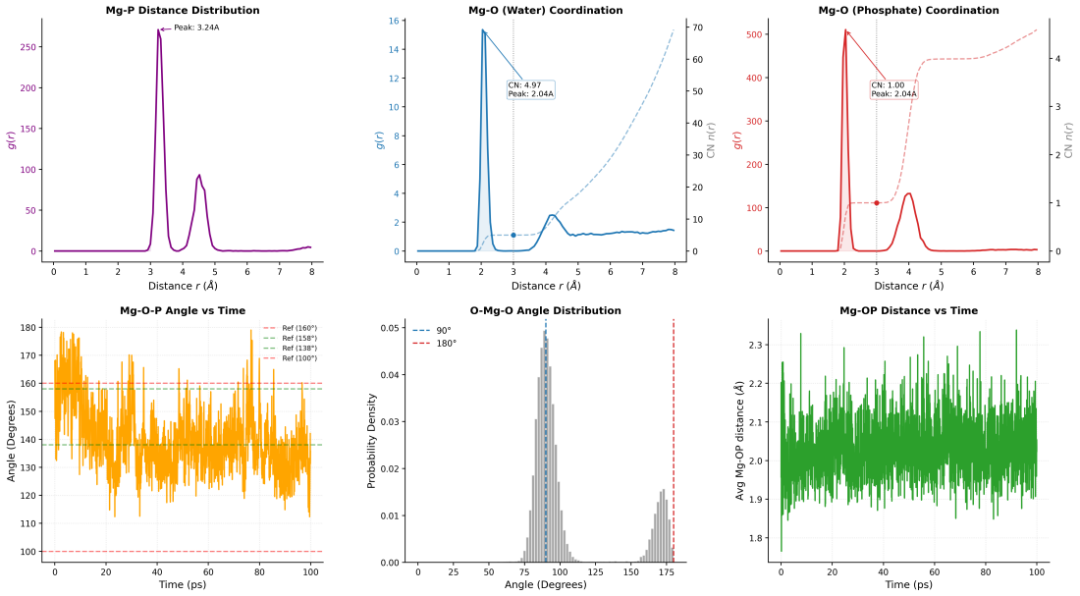
- 金属离子配位:在RNA(1L2X)与Mg²⁺的复合体系中,模型能够准确重现Mg-O键的距离分布与角度分布,体现了其对金属离子配位几何的精细刻画能力。这种能力对于研究核酸结构的稳定性与功能至关重要。
推理吞吐:在1k–50k原子范围实现约4倍提升
如何兼顾精度与效率?团队在单张NVIDIA H100 GPU上,对 MolFM-S3 与 UMA-S/M、MACE-OMol、eSEN、E2Former-V1 等多个等变模型进行了推理吞吐测试(1k–100k原子规模,保守力计算)。结果显示:
- 在1,000原子规模下,MolFM-S3 的吞吐为 61 steps/s,优于 UMA-S 的16、MACE-OMol 的8 和 E2Former-V1 的12。
- 在10,000原子规模下,MolFM-S3 仍能保持 6.10 steps/s,而多种对比模型已出现内存溢出(OOM)。
- 在1k至50k原子范围内,MolFM-S3 相对于 UMA-S 实现了约 4倍 的吞吐提升。
- 模型在单卡上最高可实现约 15万原子 的推理。然而,在100k原子规模下,由于显式建模长程作用带来的显存开销,UBio-MolFM 也遇到了瓶颈,这指明了下一步的优化方向:探索更高效的长程相互作用建模方式。
| 原子数 |
UMA-S |
UMA-M |
eSEN-30M |
MACE-OMol |
E2Former-V1 |
MolFM-S3 |
| 1,000 |
16.00 |
3.00 |
1.70 |
8.00 |
12.00 |
61.00 |
| 10,000 |
1.60 |
0.20 |
OOM |
OOM |
1.20 |
6.10 |
| 50,000 |
0.20 |
OOM |
OOM |
OOM |
OOM |
0.72 |
| 100,000 |
0.10 |
OOM |
OOM |
OOM |
OOM |
OOM |
综合来看,UBio-MolFM 的定位更接近于一个“生物体系高精度模拟底座”。它一方面将可验证的生物体系模拟推进到了更真实的原子尺度(1,300–1,500原子),另一方面通过UBio-Mol26数据与E2Former-V2架构的协同设计,使模型能够同时处理生物大分子结构、溶剂化效应与金属配位等关键相互作用,从而有望为药物发现、蛋白质构象动力学、核酸功能研究等领域提供一个统一的、更高精度的建模基础。随着其模型权重与代码的后续开源,社区将能更深入地评估和应用这一进展。
本文涉及的技术细节、评测数据及后续更新,可在云栈社区的技术文档板块持续关注与讨论。
 发表于 2026-3-5 06:13:22
|
查看: 210|
回复: 0
发表于 2026-3-5 06:13:22
|
查看: 210|
回复: 0
